New World - Living and saving Lives
Starting May 26, 2020, the Medical Device Regulation (MDR) was expected to apply in the European Union. Due to COVID-19 it was postponed by one year.
However, after years of discussion, the new framework will be a reality soon. And manufacturers, government agencies, regulatory bodies, and consumers must conform with it. Where do we stand? Does the MDR increase patient safety? Or is the innovative power of the industry weakened? And: What do we do now? An exclusive B. Braun survey at all levels of the health care system: One reform. Seven stations. Seven opinions.
Station 1: The Idea

Andreas Schwab has been a member of the European Parliament since 2004. And the CDU politician has been dealing with the MDR for almost the entire time. “The pressure on politicians was enormous,” says Schwab, “and the media coverage was quite biased. It has made our work difficult.” In May 2017 the MDR was adopted. The three-year transition period for the first device classes is expected to end. But Andreas Schwab knows that the topic will keep him busy for a long time to come.
„In 2010, defective breast implants from the manufacturer Poly Implant Prothèse caused an uproar in France. The way this company conducted business did not comply with the regulations, even given the context of that time. However, the so-called PiP scandal made it clear that there was a lack of efficient controls. For the first time, the MDR now sets a uniform application of regulations in Europe. There are also unannounced inspections. This increases patients’ safety. I took over the opinion statement for MDR from a French colleague in the European Parliament's Committee on Internal Market and Consumer Protection - I spoke a lot with companies and scientists from my electoral district and invited the Commission to southwest Germany. So, I can understand very well that some manufacturers are complaining because they have to prove the effectiveness and safety of existing devices. Even though there have only been problems with a few black sheep. We must ensure that all products meet the most modern standards in order to guarantee the best possible patient safety. Some time ago I raised concerns that the European Parliament was too ambitious in terms of the deadlines for implementation. But this has since been improved. What we need for the implementation of the MDR is a combination of orderliness and speed.“
Station 2: The Strategy

At the end of 2019, the European Commission postponed the deadlines for which so-called Class 1R devices, such as reusable surgical instruments, must be certified according to the new regulation. But does that help the manufacturers? The B. Braun Aesculap Division has been manufacturing medical devices in Tuttlingen since 1867. B. Braun board member Dr. Joachim Schulz explains why there is no way back.
„The MDR has clear advantages for patients: For example, I think it’s true that manufacturers can no longer merely rely on the equivalence principle for the approval of class III devices such as implants, they must carry out new clinical trials. However, if we have to prove that a design for surgical scissors, some of which we have been producing and supplying for over 100 years, can be sterilized through steam sterilization, unnecessary resources are tied up and our innovative strength is reduced. The MDR has created a new world. 50 to 60 percent of our employees in the development and regulatory affairs departments are currently engaged in maintaining documentation for existing devices. As a large company, B. Braun at least has the opportunity to perform many of the tests now required in-house. And we do not intend to rest on our laurels, instead we to look to the future. Technologies are changing rapidly: developments such as effective surfaces that prevent inflammation, or the robotization of the operating room, hold enormous potential. Honestly, we'd rather invent groundbreaking devices than pore over files.“
Station 3: On the Test Bench

The MDR not only changes the business of medical device manufacturers: all the notified bodies will also lose their designation under the old EU MD Directive. Whereas there were still 83 notified bodies throughout Europe in 2012, there are currently only ten. And this number cannot be suddenly increased, because the application process takes a year and a half, as Dr. Bassil Akra, Vice-President Global Strategic Business Development at TÜV SÜD in Munich, explains.
„Since 2015, at TÜV Süd we have increased our capacity by 20 percent every year. Nevertheless, we are not able to process all the requests from manufacturers for MDR certification right away. Nevertheless, I do not currently see any emergency for the availability of medical devices, as most manufacturers have so far concentrated on extending the certifications of their devices until 2024. So, the actual MDR work is still ahead of us. In principle, I think that increased monitoring and a harmonization of the legal situation that the MDR stipulates makes a lot of sense. However, the 175-page long regulation has many ambiguities and leaves room for interpretation. For example, it states that notified bodies must provide for “sufficient surveillance systems”. But what does “sufficient” really mean? So far, 32 Guidance Papers have been published with which the European Commission intends to clarify its position. Another 30 will follow by May 2020. And then, of course, we have to say that 100% harmonization will never be achieved in Europe. The languages, cultures, and legal traditions of the member states are simply too different. If, for example, the regulation states that the investigators of the notified bodies must have ‘clinical expertise’, then in Germany we understand this to mean a physician with a qualification as a specialist. In other countries, it is enough for examiners to have a bachelor’s degree in nursing or, to put it bluntly, just to have worked in a clinic. You may not like it. But that is part of the diversity in today’s united Europe.“

Station 4: The Internal Processes

The Hospital Care division of B. Braun alone has 40 locations. Medical devices are manufactured at 18 of them. Jürgen Heil, Senior Vice President Quality & Environment Management, explains how the MDR changes complex production processes.
„On a blank sheet of paper, we completely redesigned the quality processes and technical documentation from clinic, development, production and lifecycle management. This was a mammoth project, which in our Hospital Care division alone has employed more than 100 experts from all functions for several years: Quality Management, Regulatory Affairs, Development, Production, Medical Scientific Affairs, Legal, IT, Marketing, and Sales. The MDR makes so many new demands that we had to question all our external and internal processes and reorganize them, when necessary. We were able to complete some tasks very quickly: For example, the quality management certification according to the latest standard (ISO 13485:2016) at the end of 2018. One job that still keeps us occupied is that every product, in addition to the medical device label, must also be provided with a Unique Device Identification Code that is displayed in a European database (EUDAMED). However, this database will not be available for the foreseeable future. For more than 140 product families, such as IV sets or intravenous catheters, we had to create technical documentation and validation evidence and make them available in a database according to a standardized procedure. We are talking about up to 140 documents per device. But because we have been working intensely on this topic since the end of 2016 – for example, we scheduled resources at our Notified Body one year ago – we successfully completed the MDR audit last year in September. We are well positioned and can reassure our customers, who contact us with questions about availability and certification. However, we had to take some devices and sets out of the portfolio. For products from external manufacturers, for example, under the new regulation we must prove that we have access to their technical documentation, which is often not permitted at all.“
Station 5: The Market

The Prospitalia purchasing association represents more than 1,000 medical institutions, with an annual procurement volume of 1.7 billion euros. To reduce uncertainty in clinics, pharmacies, and retirement homes, the company already conducted a large-scale survey of all its medical device suppliers in 2019. One of the findings was that almost two thirds of the manufacturers already work with an MDR-accredited notified body. Here, Katja Winkels zur Strassen, Business Consultant at Prospitalia, tells us how to assess the situation there.
„The world will not end on May 26, 2020. The EU Commission has ensured this by extending the transition periods for most medical devices. But even beyond that, we have found that there is little reason for alarm. Just a year ago, however, many of our contacts on the hospital and supplier side were not well informed about MDR. Many did not know, for example, if products that were already stored in clinics could still be used when the CE expired. If it makes sense to stock up on disposable products such as catheters, for example. The answer: Yes, because as soon as a product reaches the hospital and is thus available to the user, it is considered 'put into operation'. It is a special case when products, especially large devices, are only assembled on site: commissioning only takes place when assembly is complete. As a purchasing association, we stand between manufacturers and consumers, so have a kind of turnstile function. In order to bring more transparency to the market, we conducted a survey in summer 2019 and sent it to 180 manufacturers who supply us: 64 percent - measured by the procurement volume of the affiliated institutions - worked with notified bodies that are already MDR-accredited. And at least eight percent plan to take devices off the market. We also assume that there will be a market consolidation. However, this may well make sense. For example, some doctors work with outdated devices out of habit, although there are newer alternatives that are more tolerable for patients: heavyweight and small pore hernia mesh, for example, trigger foreign body reactions more frequently than modern devices. In the case of absolute niche devices, such as those found in pediatric care, for example, it is possible for the legislature to ensure availability through a national exemption. An exciting question will be how the costs develop. According to our survey, hardly any manufacturers are planning to raise prices. But the costs for the certification will be doubled by the MDR.“
Station 6: The Medium-sized

The MedTech industry in Germany is characterized by medium-sized companies. 93 percent of the companies have less than 250 employees, while 30 percent have fewer than ten employees. The Medical Mountains medical technology cluster from Tuttlingen works with many of these small companies and start-ups and helps them to prepare for the MDR, as Managing Director Yvonne Glienke reports.
„If you hit the streets in Tuttlingen today as a regulatory affairs expert, you can’t escape getting job offeres. Small manufacturers especially have problems attracting the skilled workers they need. At Medical Mountains, we organized a total of 94 seminars with over 1000 participants in 2019. The focus was clearly on topics such as quality management and certifications. The MDR is leading to a consolidation in the market. Small companies in particular are increasingly saying ‘no’ to the question of whether the effort is worthwhile – they are joining forces, selling, or changing their business model and operating as an extended workbench for their major customers. We have been involved with the MDR for over a decade, we have spoken with politicians, written position papers – and we continue to fight for more practical application. For example, why do I need a paper instruction manual in all EU languages for every disposable scalpel? I could also imagine that some companies launch their innovative devices in the USA or China first, to circumvent the MDR. But the MDR has one advantage in any case: cooperation between companies in the region is greatly improved. There is less competition and more collaboration. This might lead to something new.“
Station 7: The Human Factor

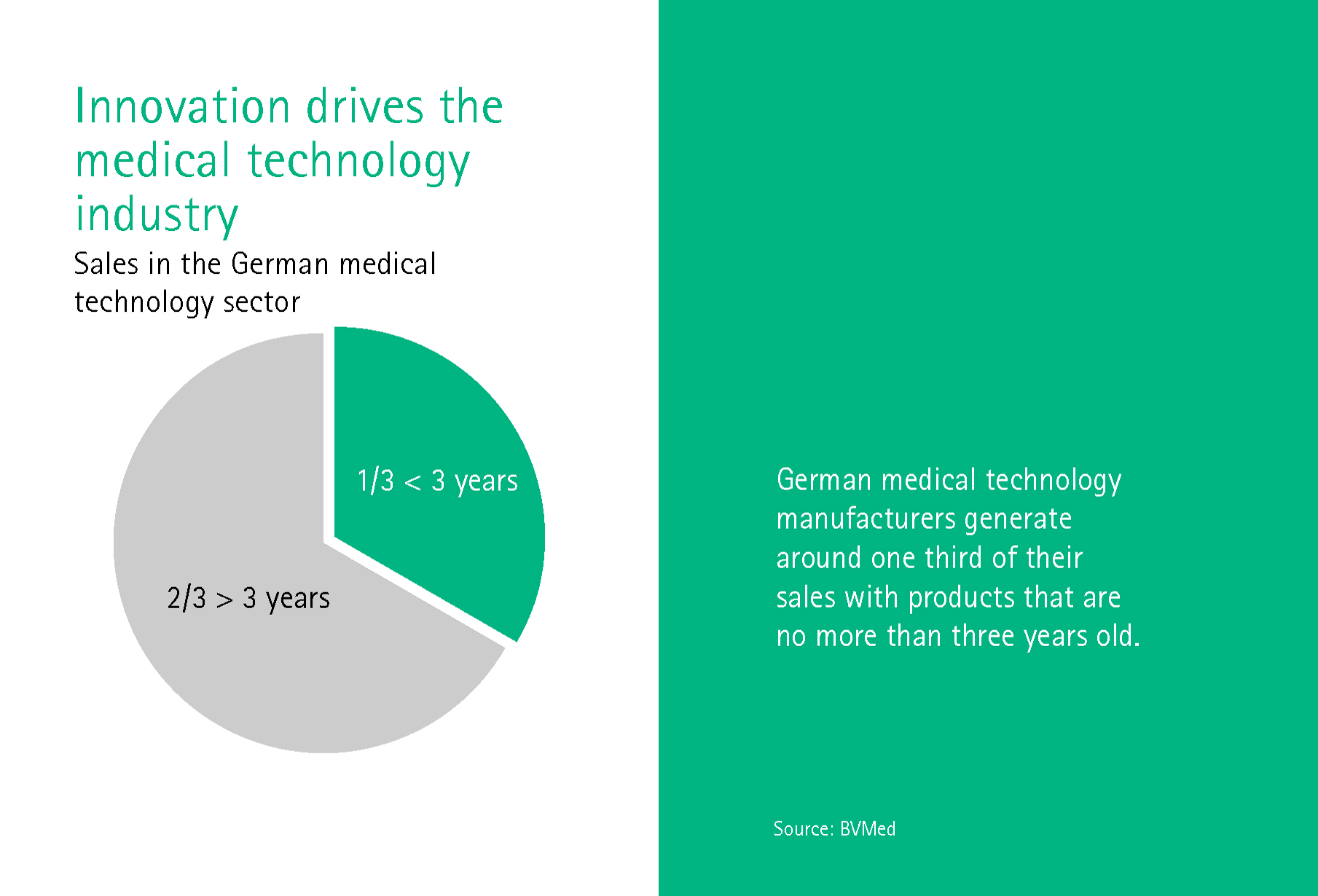
Dr. Giovanni Rubeis works at the Institute for History and Ethics of Medicine at the University of Heidelberg and is primarily concerned with the ethical aspects of digital transformation. What should a legal framework that keeps pace with change look like, asks Rubeis? After all, the MedTech industry makes a third of its turnover with devices that are no older than three years.
„The healthcare system is changing rapidly: currently, for example, there are 10,000 psychotherapy apps available to download. But are these actually medical devices? And how are the new AI applications, which seemed unthinkable a few years ago, classified and regulated? The economy turns knowledge into devices so quickly that the legislator can hardly keep up. The technological transformation will also put the MDR to the test. The interest of the patient is very clear: the device must help me. And it must be safe. From an ethical point of view, it should therefore be welcomed that the regulatory framework has been tightened and clarified. What gets me thinking is that the additional expenditure should be met with similar resources and staffing levels. It is not enough to pass a law. The appropriate infrastructure and resources must also be provided. The MDR is a very complex affair. And this is also an ethical problem. Because it is not easy for patients and even for medical professionals to find their way around. It is true that all information should be accessible in the EUDAMED database. But it won't be ready until 2024, and it is mainly aimed at experts. Of course, most people trust their doctor. But a regulation is not enough to increase the lost confidence in the medical devices sector. Politics and business must consider how to communicate with patients and provide all the information needed to make a decision themselves.“